Msuite2:一体式DNA甲基化分析及可视化软件
什么是DNA甲基化
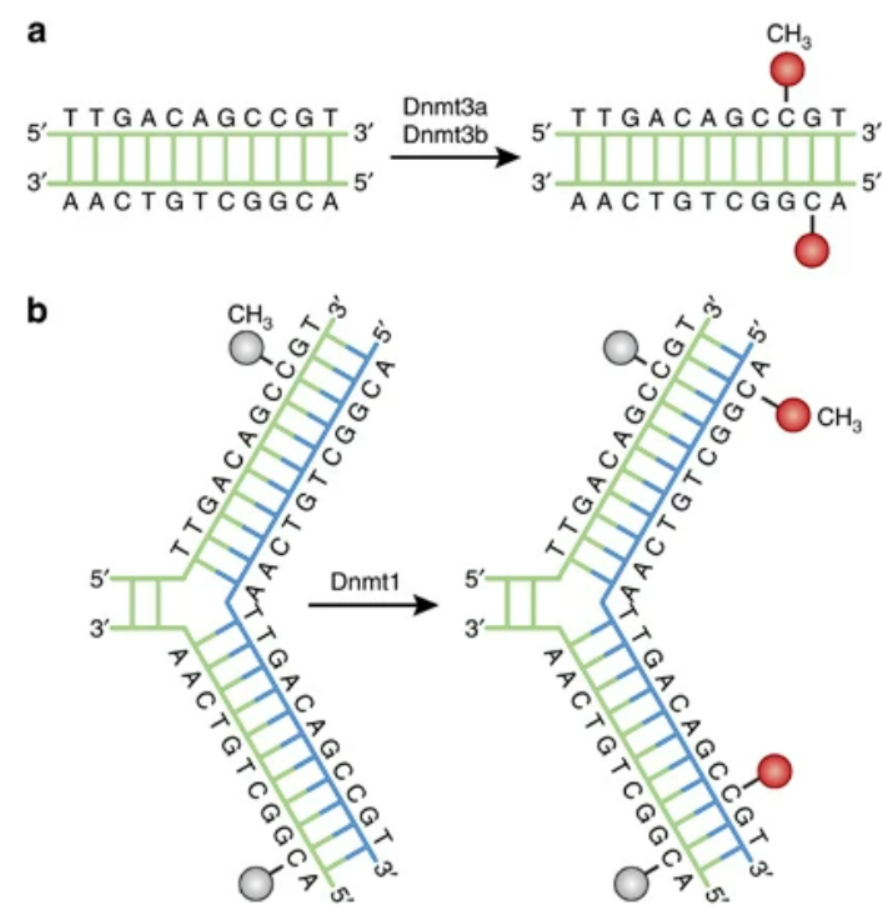
DNA甲基化是一种重要的表观遗传调控因子。在哺乳动物基因组中,最普遍且重要的DNA甲基化类型是5-甲基胞嘧啶(5mC)。该类型会在DNA甲基转移酶(DNMT)的作用下,将S-腺苷甲硫氨酸(S-adenyl methionine)的甲基基团转移到胞嘧啶的第五个碳原子上,从而形成5-甲基胞嘧啶(5mC)[1,2]。这种现象主要发生在CpG二核苷酸位点上,即一个胞嘧啶(C)核苷酸与一个鸟嘌呤(G)核苷酸相邻的位置。
DNA甲基化与癌症
众所周知,DNA甲基化模式的改变往往和癌症相关。正常个体基因组中大部分CpG位点都是存在甲基化的,而CpG岛以及远端的调控元件区域则是对DNA甲基转移酶(DNMT)具有一定抗性。而在癌症细胞中,基因组上的5mC会出现大范围丢失,而在启动子或者增强子等区域会出现异常增加。而正是这种DNA甲基化模式的改变,抑制了抑癌基因的表达并且促进了原癌基因的表达,从而促进了肿瘤的发生[1]。
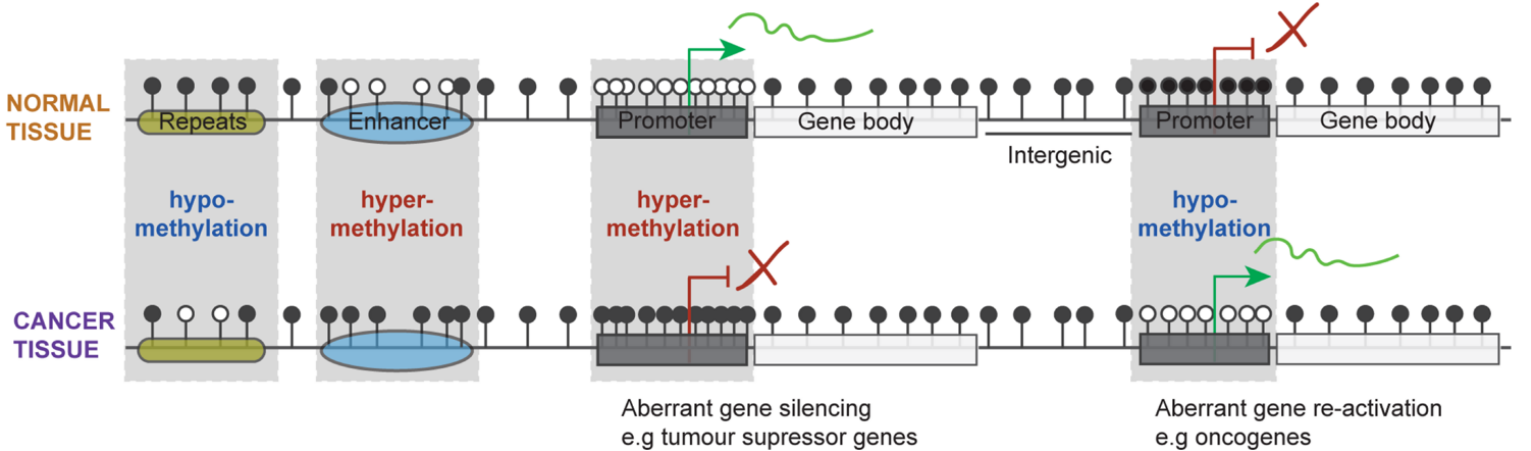
白色圆圈代表未甲基化CpG位点;黑色圆圈代表甲基化CpG位点
DNA甲基化的分析手段
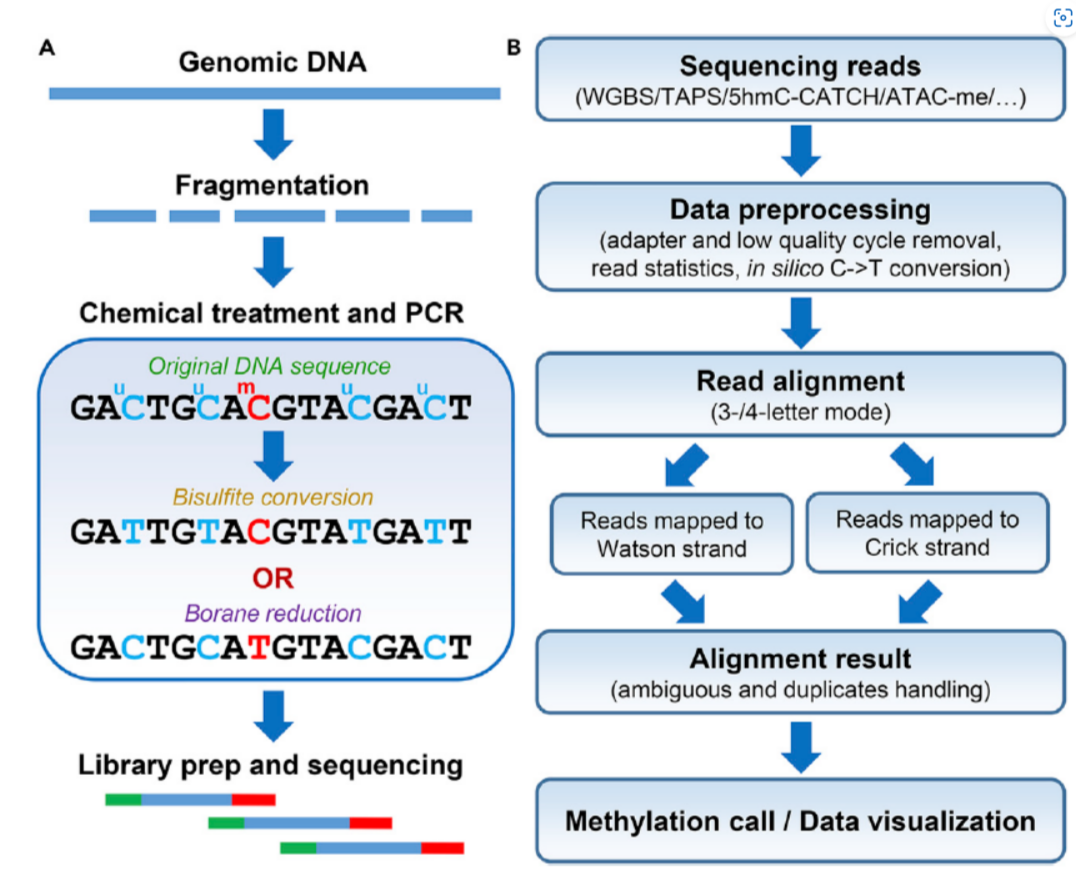
在过去几年中,已经有多种高分辨率的DNA甲基化分析方法被开发出来。其中最常见的方法是重亚硫酸盐法[6,7]。为了区分甲基化的胞嘧啶和非甲基化的胞嘧啶,该方法将未甲基化的胞嘧啶转化成尿嘧啶,而经过甲基化修饰的胞嘧啶并不受影响,仍然保持胞嘧啶的状态。随后在进行PCR进行扩增时,尿嘧啶会被识别为胸腺嘧啶。随后在PCR的扩增下,序列中未甲基化的C都变成了T。由于在哺乳动物中被甲基化修饰的胞嘧啶主要出现在CpG二核苷酸位点上,仅占了全基因组C的5%左右。因此这种方法会导致基因组大部分胞嘧啶发生改变,从而使得测序数据的GC含量与真实含量相差很大。
为此研究人员开发了非重亚硫酸盐法来检测DNA甲基化(例如TAPS法[5])。与重亚硫酸盐法相比,该方法只会对将甲基化的C转化为T,而未甲基化的C不会改变。这样就使得只有很少的胞嘧啶发生的改变,从而解决GC偏差问题,并且还可以获得更高的序列复杂性和更低的DNA降解率等优势。
Msuite的技术优势
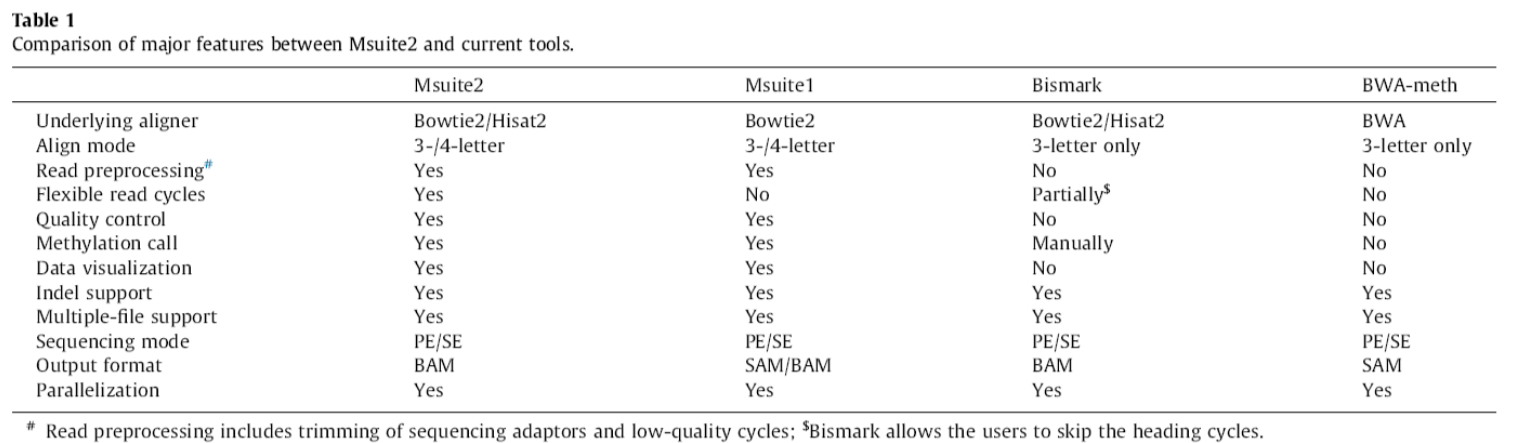
然而,目前主流的DNA甲基化分析工具大多只支持重亚硫酸盐法的数据处理,而对于非重亚硫酸盐法所产生的数据并不兼容。部分软件支持非重亚硫酸盐法的数据处理,但是这些软件大多仅使用的是通配符比对法,导致其比对效率以及准确度较低。除此之外,市面上绝大多数软件仅仅只支持序列比对,在上游质控、下游甲基化位点检测以及数据可视化等方面并不完善。在此,我向你推荐一款一体式DNA甲基化分析及可视化软件:Msuite[3,4]。该软件兼容当前市面上所有主流的DNA甲基化数据。同时软件还集成了质控、比对、甲基化位点检测以及数据可视化一体式服务,真正做到了性能出众,易于使用。
Msuite2方法
搭建运行环境及其依赖库
首先我们下载最新的Msuite2的程序包 ,并通过tar zxf Msuite2-2.1.0.tar.gz进行解压。其中主程序msuit2就在新创建的文件夹msuite2-2.1.0中。
为了顺利的运行Msuite2,您还需要确保您的Linux/Unix系统中安装了g++ (v4.8)、perl (v5.10)、R (v3.0)、Bash 4,并且保证其版本号不低于上述所提到的版本号。
此外您还需要安装bowtie2、hisat2、samtools,并且确保这些软件在您的环境变量中可以直接唤起。
当然msuite2-2.1.0文件夹中还包含了预编译的可执行文件。如果您的主程序msuite2不能正常使用,或者您想让msuite2更加适配您的系统,您可以运行如下命令:
1 | user@linux$ make clean && make |
注意: 大部分主程序
msuit2不能运行都是由于较低的libc++库导致的。
构建索引
在运行Msuite2时,您需要先对参考基因组构建索引。为此,您只需要准备GENOME.FA、REFSEQ.txt便可以使用./msuite2-2.1.0/build.index/build.index.sh来进行构建。
1 | ./msuite2-2.1.0/build.index/build.index.sh \ |
其中hg38和mm10的RefSeq文件都已经下载在./msuite2-2.1.0/build.index/文件夹中,您也可以从UCSC genome browser下载其他物质或其他基因版本的RefSeq文件。
值得注意的是:
- 对于基因序列文件
GENOME.FA,您需要提供包含多个fasta的单一文件,或者提供包含多个染色体序列文件的文件夹; build.index.sh会自动整合Lambda基因组来构建索引;RefSeq.txt可以使用GFF格式的注释文件(注意:文件名必须包含gff字符);- 此外
GENOME.FA、RefSeq.txt(orGene.anno.gff)支持Gzip或者Bzip2压缩格式。注意,压缩文件后缀必须与压缩格式相对应(例如:Gzip压缩格式的REFSEQ.txt.gz文件以及Bzip2压缩格式的REFSEQ.txt.bz2文件); Genome.ID是构建基因组索引的名称,并会写入到./msuite2-2.1.0/index/当中。并且,构建基因组索引的个数不受限制。
运行Msuite2
您可以通过运行下述命令来将Msuite2添加到您的环境变量中:
1 | export PATH=$PATH:$PWD |
Msuite2提供了两种分析模式3-letter和4-letter模式。如果您的数据是重亚硫酸盐法,您需要选择3-letter模式,并且需要设置-m BD参数;如果您的数据是非重亚硫酸盐法或者其他non-CpG甲基化比例很低的数据(例如,人体的大部分体细胞组织)时,您需要选择4-letter模式并设置-m TAPS参数。3-letter模式:
1 | msuite2 -x hg38 -m BS -3 -p 8 \ |
4-letter模式:
1 | msuite2 -x hg38 -m TAPS -4 -p 8 \ |
在运行上述命令后,makefie 文件会被生成在输出文件夹中(-o选项)。此时您可以在该文件中运行如下命令:
1 | cd /path/to/output/dir; make |
然后Msuite2会对数据进行处理,包括去接头,质控,比对,甲基化分析以及数据可视化。
值得注意的是:
-x指定参考基因组索引,与之前的Genome.ID相对应;-m参数与-3/-4必须相互对应。对于WGBS数据,可以设置-m BS和-3参数,对于TAPS数据或类似数据,可以设置-m TAPS和-4参数;-p是确定使用线程数,-p 0设置为使用所有线程数;-1和-2指定输入文件,如果您的输入序列为单链只需提供-1参数即可,-o指定输出文件;- 您可以通过设置
-k nextera来直接分析ATAC-me或类似数据; - 如何您需要添加多个
.fq文件,您可以通过,来进行分割。例如:-1 ./lane1.read1.fq.gz,./lane2.read1.fq.gz -2 ./lane1.read2.fq.gz,./lane2.read2.fq.gz - 如果您需要添加所有
.fq文件,您可以按照如下来设置-1 '/path/to/lane*.read1.fq.gz' -2 '/path/to/lane*.read2.fq.gz',请注意单引号对于防止*****被shell提取十分重要; - 如果您不想分析完整reads,您还可以通过设置
-c cycle参数来实现。例如:您由于序列质量问题只想分析reads的前75bp,您可以设置-c 75参数。如果您想要跳过头尾的5/10 bp,您可以设置--cut-r1-head 5 --cut-r1-tail 10 --cut-r2-head 5 --cut-r2-tail 10来实现;
此外,您还可以在./msuite2-2.1.0/testing_dataset中找到测试文件进行测试。并且Msuite2还提供了./msuite2-2.1.0/run_testing_dataset.sh作为示例,用于构建引用并自动在测试数据集上运行分析。用户可以使用以下命令调用它:
1 | ./run_testing_dataset.sh |
其他功能
并且Msuite2还包括一个Mviewer工具来提供核苷酸水平,genotype-preserved DNA methylation数据的可视化。更多信息可以参考Mviewer文件夹中的README文件。
此外,./msuite2-2.1.0/util/文件夹中,您可以使用pe_bam2bed.pl和se_bam2bed.pl程序将BAM文件转化为BED格式文件,还可以通过bed2wig来将BED文件转化为WIG文件。此外您还可以使用extract.meth.in.region来提取特定区域的CpG位点,C-count和T-count。
Msuite2结果分析
Msuite运行的所有结果都放在-o OUTDIR所指定的文件夹中,同时Msuite会生成HTML文件(Msuite2.report/index.html)来报告分析结果质量:包括质控情况,比对率,CpG位点的总体甲基化水平,M-bias,转化效率(通过比对到Lamda基因组的reads数来估计)
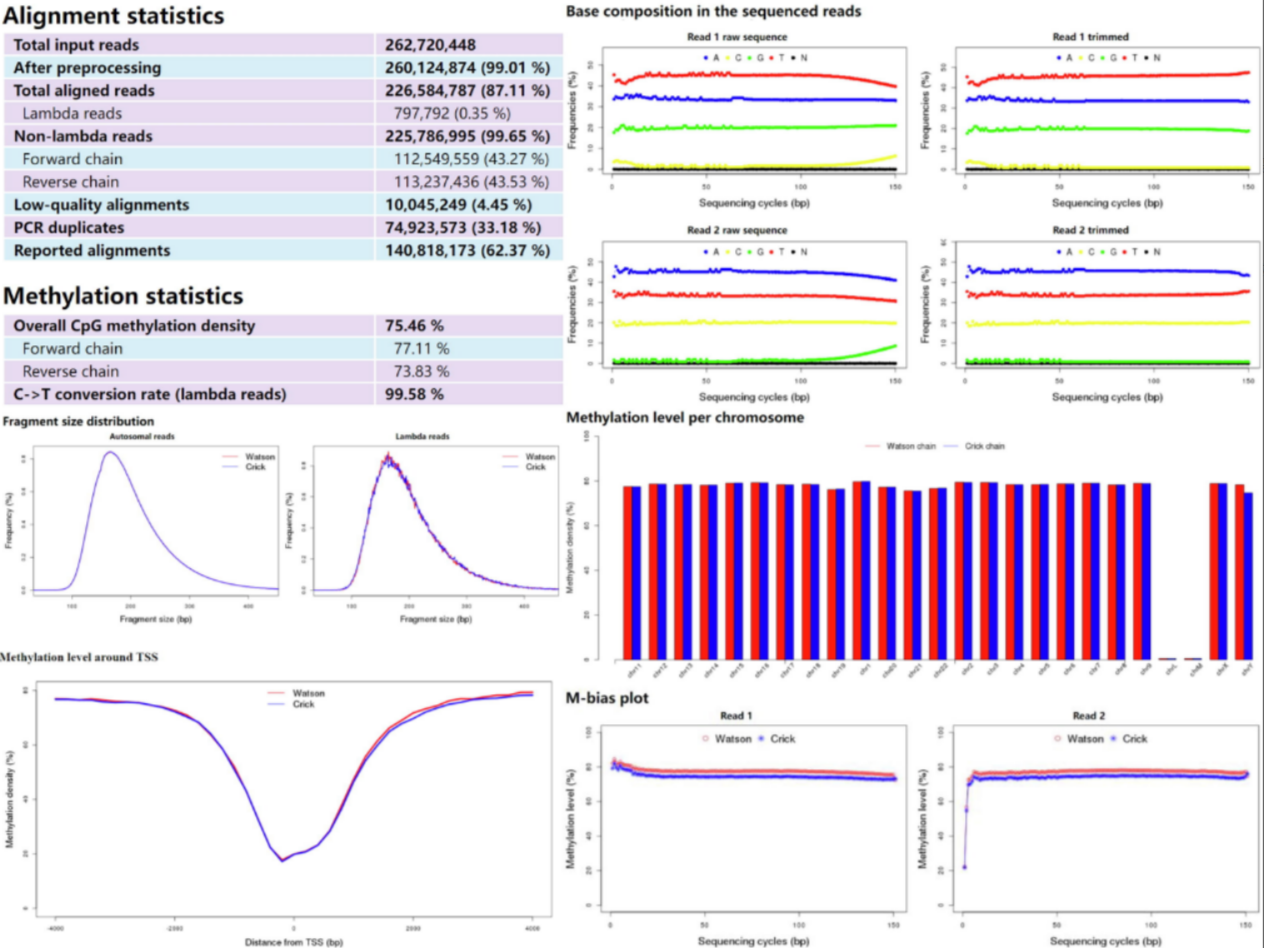
比对结果被存储在bam文件和sam文件中:Msuite2.final.bam和Msuite2.final.sam。甲基化结果被存储在Msuite2.CpG.meth.call,Msuite2.CpH.meth.call和Msuite2.CpG.meth.bedgraph。
您可以在输出文件中运行make clean来删除中间文件以节省存储空间。
- Skvortsova, Ksenia, Clare Stirzaker, and Phillippa Taberlay. “The DNA methylation landscape in cancer.” Essays in biochemistry 63.6 (2019): 797-811.
- Moore, Lisa D., Thuc Le, and Guoping Fan. “DNA methylation and its basic function.” Neuropsychopharmacology 38.1 (2013): 23-38.
- Li, Lishi, et al. “Msuite2: All-in-one DNA methylation data analysis toolkit with enhanced usability and performance.” Computational and Structural Biotechnology Journal 20 (2022): 1271-1276.
- Sun, Kun, et al. “Msuite: a high-performance and versatile DNA methylation data-analysis toolkit.” Patterns 1.8 (2020): 100127.
- Liu, Yibin, et al. “Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution.” Nature biotechnology 37.4 (2019): 424-429.
- Cokus, Shawn J., et al. “Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning.” Nature 452.7184 (2008): 215-219.
- Lister, Ryan, et al. “Highly integrated single-base resolution maps of the epigenome in Arabidopsis.” Cell 133.3 (2008): 523-536.
Msuite2:一体式DNA甲基化分析及可视化软件